
test génétique pour le
planning familial
Dépistage de + 2 000 maladies héréditaires
Pour CHF 1 495
Depuis votre canapé.
Pourquoi faire le test ?
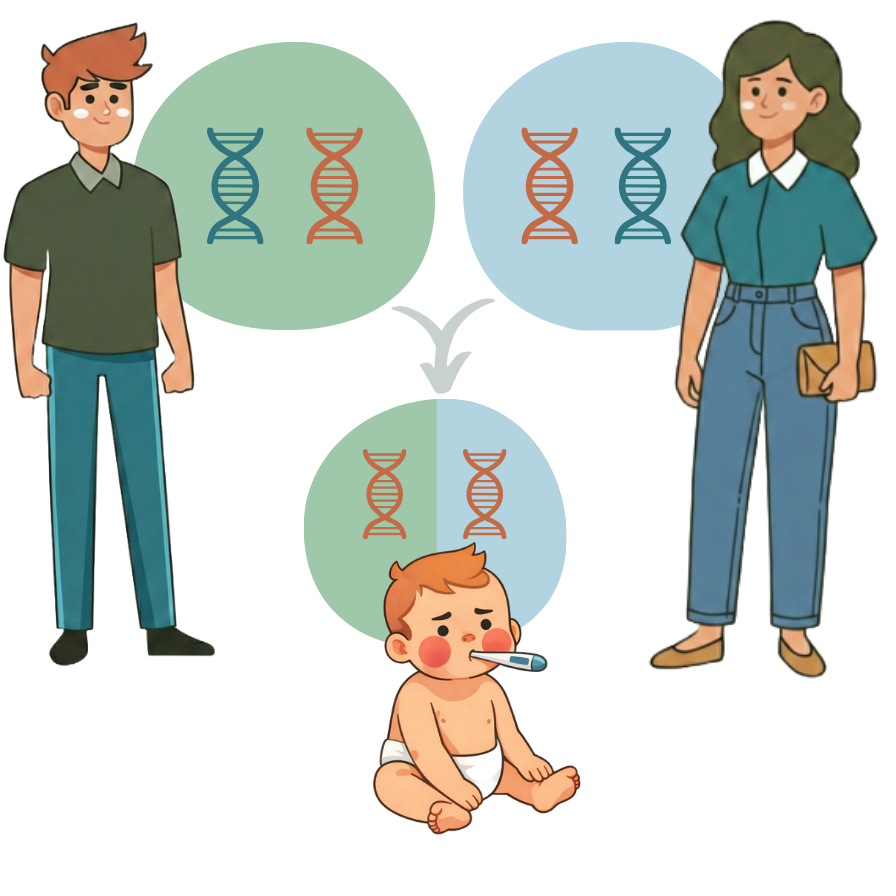
Cela surprend souvent les futurs parents, mais la plupart des bébés nés avec certains types de maladies génétiques ont des parents en bonne santé sans antécédents familiaux évidents de la maladie. De nombreuses personnes sont porteuses de mutations génétiques sans le savoir. Les mutations génétiques peuvent rendre un gène défectueux. Comme nous avons deux copies de la plupart des gènes (une de chaque parent), une copie saine compense généralement la copie défectueuse. Cependant, si les deux parents sont par hasard porteurs de la même maladie récessive, il existe un risque de 25 % que leur enfant hérite des deux copies défectueuses et soit atteint.
Des études à grande échelle ont établi à quel point ces maladies génétiques héréditaires sont fréquentes dans la population générale. Dans une étude américaine portant sur des centaines de milliers de personnes, la proportion estimée de grossesses touchées était d’environ 1 sur 175 [1]. De même, dans un récent programme national australien de dépistage portant sur plus de dix mille couples, environ 1 couple sur 50 a été identifié comme présentant un risque accru d’avoir un enfant atteint [2].
- Westemeyer M, Saucier J, Wallace J, et al. Clinical experience with carrier screening in a general population: support for a comprehensive pan-ethnic approach. Genetics in Medicine. 2020;22:1320–1328. doi:10.1038/s41436-020-0807-4
- Mackenzie's Mission Investigators. Nationwide implementation of reproductive genetic carrier screening in Australia. New England Journal of Medicine. 2024. doi:10.1056/NEJMoa2314768
Le test
Le dépistage de porteur élargi est un test basé sur l’ADN qui analyse le matériel génétique d’une personne afin de déterminer si elle est porteuse de certaines maladies héréditaires. Grâce à la technologie de séquençage de nouvelle génération, ce type de dépistage peut examiner simultanément des centaines, voire des milliers de gènes. Le test analyse spécifiquement 1 944 gènes associés à des maladies graves qui touchent généralement les enfants tôt dans la vie. Le processus est simple et non invasif et repose sur un échantillon de salive contenant de l’ADN pouvant être extrait et séquencé. Le test se concentre sur les maladies récessives et liées à l’X — des maladies génétiques qui ne provoquent généralement pas de symptômes chez les parents, mais qui peuvent entraîner des maladies graves chez leurs enfants si les deux parents portent la même maladie ou si certains modes de transmission sont en jeu.
Le laboratoire
Nous travaillons avec CeGaT GmbH comme laboratoire partenaire pour le dépistage de porteur élargi. CeGaT réalise l’analyse génétique à l’aide d’un test développé en interne, permettant la détection de variants mononucléotidiques et de variations du nombre de copies, ainsi que des tests ciblés pour des maladies telles que le syndrome de l’X fragile et l’amyotrophie spinale. Le laboratoire dispose des principales accréditations internationales pour les tests génétiques cliniques, notamment CAP, CLIA et ISO 15189.
Conseil génétique
Le conseil génétique avant et après le test avec un·e spécialiste constitue un élément clé de notre service. Ces séances permettent de poser des questions, de comprendre la portée et les limites du test, et de discuter des résultats en détail. En examinant les antécédents personnels et familiaux, le conseil aide à replacer les résultats dans leur contexte et favorise une prise de décision éclairée.
Comment ça marche
Testez votre statut de porteur depuis chez vous.
Conseil génétique
Échangez en ligne avec un·e conseiller·ère en génétique certifié·e pour comprendre ce que signifie le dépistage de porteur pour vous et votre famille.
Prélever un échantillon
Prélevez simplement un échantillon de salive à domicile à l’aide du kit que nous vous envoyons. Aucune visite en clinique n’est nécessaire — il suffit de fermer le tube et de le renvoyer avec l’étiquette prépayée.
Discuter des résultats
Passez en revue vos résultats avec votre médecin ou notre conseiller·ère en génétique, qui vous aidera à comprendre ce que ces résultats signifient pour vos projets.
Liste des gènes
Neurology 423 gènes+
Muscular dystrophies, congenital myopathies, congenital myasthenic syndromes, hereditary motor and sensory neuropathies, epileptic encephalopathies, leukodystrophies, neuronal ceroid lipofuscinoses, hereditary ataxias, hereditary spastic paraplegias, brain malformations, peroxisomal disorders, neurodegeneration with brain iron accumulation, and other neuromuscular and neurodegenerative conditions
AAAS, AARS1, ABCD1, ABHD12, ACOX1, ACP5, ADAR, ADCY5, ADCY6, ADGRG1, ADGRG6, ADPRS, AFG3L2, AGTPBP1, AIMP1, ALDH3A2, ALDH5A1, ALG13, ALS2, AMPD2, ANO5, AP1S2, AP3B2, AP4M1, AP4S1, APC2, APTX, ARFGEF2, ARL6IP1, ARMC9, ARSA, ARV1, ARX, ASCC1, ASPA, ASPM, ATCAY, ATP13A2, ATP1A2, ATP2B3, ATP7B, AVIL, B3GALNT2, B4GALNT1, B4GAT1, BCAP31, BLTP1, BRF1, BSCL2, C19orf12, CA8, CAPN3, CASK, CCT5, CENPF, CEP41, CERS1, CHAT, CHMP1A, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, CLCN1, CLCN2, CLP1, CNPY3, CNTNAP1, COASY, COL13A1, COL6A1, COL6A2, COL6A3, COLQ, COQ4, COQ8A, COX10, CPLANE1, CRB2, CRPPA, CRYAB, CSF1R, CSTB, CTDP1, CTNNA2, CWF19L1, CYP27A1, CYP2U1, CYP7B1, D2HGDH, DAG1, DARS1, DARS2, DCAF17, DCX, DDC, DDHD1, DDHD2, DDX59, DENND5A, DGUOK, DHDDS, DLAT, DMD, DMXL2, DNAJC19, DNAJC3, DNAJC6, DOK7, DPYD, DST, DYSF, ECEL1, EGR2, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S3, ELOVL4, ELP1, EML1, ENTPD1, EPM2A, ERCC6, ERCC8, ERLIN1, ERLIN2, ETFDH, EXOC3L2, EXOSC3, EXOSC8, EXOSC9, FA2H, FAM149B1, FBXO7, FGD4, FHL1, FIG4, FKRP, FKTN, FLVCR1, FLVCR2, FOLR1, FRRS1L, FTL, FUCA1, FXN, GALC, GBA2, GBE1, GCH1, GJC2, GMPPB, GOLGA2, GOSR2, GOT2, GPAA1, GPSM2, GPT2, GRID2, GRIN1, GRM1, GRM7, GUCY1A1, GUF1, HACD1, HACE1, HADHA, HADHB, HARS1, HEPACAM, HEXA, HEXB, HIKESHI, HINT1, HPDL, HSD17B10, HSPD1, HYCC1, HYLS1, IFIH1, IGHMBP2, INPP5K, ITGA7, ITPR1, JAM2, JAM3, KCNJ10, KCNMA1, KDM5C, KIDINS220, KIF1A, KIF1C, KIFBP, KLHL40, KLHL41, KY, L2HGDH, LAMA2, LAMB1, LAMC3, LARGE1, LIMS2, LMOD3, LPIN1, LYST, MAG, MBOAT7, MECP2, MECR, MLC1, MMACHC, MPZ, MRE11, MSTO1, MTFMT, MTM1, MTMR2, MTTP, MUSK, MYBPC1, MYH3, MYMK, NARS1, NAXE, NCAPD3, NDE1, NDRG1, NDUFA12, NDUFA2, NDUFS4, NDUFS7, NDUFS8, NDUFV1, NEB, NFASC, NGF, NHLRC1, NKX6-2, NRROS, NRXN1, NT5C2, NTRK1, NUP188, OCLN, OPA3, OPHN1, ORAI1, OSGEP, PANK2, PCCA, PCCB, PCDH12, PCYT2, PDE10A, PDHA1, PEX1, PEX10, PEX12, PEX13, PEX16, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7, PI4KA, PIEZO2, PIP5K1C, PLA2G6, PLCB1, PLEC, PLEKHG5, PLP1, PMP22, PNKP, PNPLA6, PNPT1, POLG, POLR3A, POLR3B, POMGNT1, POMGNT2, POMK, POMT1, POMT2, PRDM12, PRICKLE1, PRKRA, PRPS1, PRX, PTPN23, PTRH2, PTS, PYCR2, PYROXD1, QDPR, RAB39B, RAD21, RAPSN, RARS1, RELN, RETREG1, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2, ROBO3, RRM2B, RTTN, RXYLT1, RYR1, SACS, SAMHD1, SBF1, SBF2, SCARB2, SCN1B, SCN4A, SCN9A, SCO2, SELENON, SEPSECS, SETX, SGCA, SGCB, SH3TC2, SIL1, SLC12A6, SLC16A2, SLC18A3, SLC1A4, SLC25A12, SLC25A4, SLC2A1, SLC30A10, SLC39A14, SLC52A2, SLC52A3, SLC5A7, SLC6A3, SLC6A5, SLC6A8, SLC9A1, SLC9A6, SMPD4, SNORD118, SNX14, SOD1, SPART, SPG11, SPR, SPTBN2, SPTBN4, SQSTM1, STAC3, STAMBP, STIM1, STRADA, STUB1, SUCLA2, SUOX, SURF1, SVBP, SYN1, SYNE1, SYNJ1, SZT2, TANGO2, TBC1D24, TDP2, TECPR2, TH, TIMM8A, TMX2, TNNT1, TOE1, TPM3, TRAK1, TRAPPC11, TRAPPC12, TREX1, TRIM2, TRIM32, TRIP4, TSEN2, TSEN54, TTC19, TTN, TTPA, TYMP, UBA1, UBA5, UCHL1, UFC1, UGP2, UNC80, USP18, VAC14, VAMP1, VLDLR, VMA21, VPS11, VPS13D, VPS37A, VPS41, VPS53, VRK1, WARS2, WASHC5, WDR45, WDR45B, WDR62, WDR73, WDR81, WNK1, WWOX, ZC4H2, ZFYVE26
Metabolic 286 gènes+
Inborn errors of metabolism including amino acid disorders, organic acidemias, fatty acid oxidation disorders, urea cycle disorders, glycogen storage diseases, lysosomal storage disorders, peroxisomal disorders, congenital disorders of glycosylation, cholesterol biosynthesis defects, mineral and metal metabolism disorders, and other metabolic conditions
ABCD4, ACADM, ACADS, ACADSB, ACADVL, ACAT1, ACOX2, ACY1, ADSL, AGA, AGL, AGPS, AGXT, ALAD, ALDH18A1, ALDH4A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG14, ALG3, ALG6, ALG8, AMACR, AMPD1, AMT, ANO10, ARG1, ARSB, ARSL, ASAH1, ASL, ASS1, ATIC, ATP6AP1, ATP6V0A2, ATP7A, ATPAF2, AUH, B3GALT6, B3GAT3, B4GALT1, B4GALT7, BCKDHA, BCKDHB, BCKDK, CA5A, CARS2, CBS, CHST14, CHST3, CHSY1, CLDN16, CLDN19, CLN3, CLN5, CLN6, CLN8, CLPP, CNNM2, COG1, COG2, COG4, COG5, COG6, COG7, COQ7, COX14, CPS1, CPT1A, CPT2, CTNS, CTSA, CTSD, CTSK, DHCR24, DHCR7, DHODH, DHTKD1, DNAJC12, DNM1L, DOLK, DPAGT1, DPM1, DPM2, DYM, EBP, EPG5, ERAL1, ETFA, ETFB, FAR1, FASTKD2, FBP1, FCSK, FITM2, FTCD, FUT8, G6PC1, G6PC3, GAA, GALK1, GALNS, GAMT, GBA1, GCDH, GDAP1, GFER, GFM1, GFM2, GFPT1, GK, GLB1, GLDC, GLS, GLYCTK, GM2A, GMPPA, GNPAT, GNPTAB, GNPTG, GNS, GPHN, GRHPR, GTPBP3, GUSB, GYS1, GYS2, HADH, HARS2, HCFC1, HGSNAT, HJV, HLCS, HMGCL, HMGCS2, HOGA1, HPRT1, HSD11B2, HSD17B4, HTRA2, HYAL1, IARS2, IDS, IDUA, ITPA, IVD, KCTD7, LARS2, LDHA, LDLR, LFNG, LIPA, LMBRD1, LPL, LYRM4, MAGT1, MAN1B1, MAN2B1, MANBA, MAOA, MAT1A, MCCC1, MCCC2, MCEE, MCOLN1, MFSD8, MGAT2, MLYCD, MMAA, MMAB, MMADHC, MMUT, MOCS1, MOCS2, MOGS, MPDU1, MPI, MRPL3, MRPS16, MSMO1, MTHFR, NAGA, NAGLU, NAGS, NDUFA10, NDUFAF1, NDUFAF4, NDUFS6, NEU1, NGLY1, NNT, NPC1, NPC2, NSDHL, NUBPL, OGDH, OTC, OXCT1, PAH, PARS2, PC, PCK1, PEPD, PEX11B, PEX14, PEX19, PFKM, PGAP2, PGAP3, PGK1, PGM1, PGM3, PHGDH, PHKG2, PHYH, PIGA, PIGL, PIGN, PIGO, PIGS, PIGT, PIGV, PMM2, PNPO, POR, PPT1, PRODH, PSAP, PSAT1, PYCR1, PYGL, PYGM, RBCK1, RFT1, RMND1, SAR1B, SC5D, SEC23B, SGSH, SLC12A3, SLC16A1, SLC17A5, SLC22A5, SLC25A13, SLC25A15, SLC25A20, SLC25A22, SLC25A42, SLC2A2, SLC35A1, SLC35A2, SLC35C1, SLC35D1, SLC37A4, SLC39A8, SLC3A1, SLC46A1, SLC5A1, SLC7A7, SMPD1, SSR4, ST3GAL3, STT3A, SUCLG1, SUMF1, TACO1, TMEM165, TMEM70, TPP1, TRMT10C, TRPM6, TUFM, TUSC3, UMPS, UQCRB, UROS, VMA12, VMA22, VPS33A, XYLT1, XYLT2
Developmental 179 gènes+
Syndromic and non-syndromic intellectual disability, global developmental delay, autism spectrum-associated syndromes, brain malformations, GPI anchor deficiency syndromes, DNA repair and chromosome maintenance disorders, and other neurodevelopmental conditions
ABCC9, ACACA, ACO2, ACSL4, ACTL6B, ADAM22, ADARB1, AFF2, AHCY, AIMP2, ALG9, AP4B1, AP4E1, ARHGEF9, ATAD1, ATG7, ATP6AP2, ATP8A2, ATRX, B9D1, BRWD3, CAD, CAMK2A, CC2D1A, CCDC22, CCDC88A, CCDC88C, CDH11, CEP55, CHKB, CIT, CKAP2L, CNKSR2, CNTNAP2, CPLX1, CRADD, CRBN, CTU2, CYB5R3, DBT, DCHS1, DDX11, DDX3X, DEAF1, DEGS1, DHX37, DLG3, DOCK7, EIF4A3, EMC1, EMC10, EPRS1, ERCC1, EXT2, EXTL3, FOXL2, FOXRED1, GAD1, GAN, GATM, GCSH, GDI1, GEMIN4, GLUL, GRIA3, GRIK2, HERC2, HOXA1, HPD, HUWE1, IARS1, IGF1, IL1RAPL1, INTS1, IQSEC2, KATNIP, KDM5B, KIF14, KLHL7, KNL1, KPTN, L1CAM, LAMA1, LAS1L, MED12, MED17, MED25, METTL5, MFSD2A, MRPS34, MSL3, MTR, NADSYN1, NALCN, NDST1, NECAP1, NEMF, NEXMIF, NKAP, NONO, NSUN2, NUDT2, OTUD5, OTUD6B, OXR1, PAM16, PCDH19, PHF6, PIGB, PIGG, PIGK, PIGP, PIGQ, PLAA, PLEKHG2, PLVAP, PNPLA8, POLA1, POLR1C, PPP1R21, PQBP1, PRUNE1, PTCHD1, PUS7, QARS1, RALGAPA1, RDH11, RINT1, RLIM, RNF113A, RNF13, RPIA, RPS6KA3, RTN4IP1, RUSC2, SCAPER, SCYL1, SDHAF1, SLC12A5, SLC13A5, SLC25A1, SLC5A6, SLC6A9, ST3GAL5, STIL, TAF1, TAF2, TAF6, TASP1, TBC1D23, TBCD, TCTN3, TELO2, THOC6, TIMM50, TMEM132E, TMEM260, TMEM94, TMTC3, TRAIP, TRAPPC4, TRAPPC9, TRIT1, TRMT1, TUBGCP2, TUBGCP6, UBE2A, UFM1, UGDH, UROC1, WDR4, WNT1, XRCC4, YIF1B, ZBTB24, ZC3H14, ZDHHC9, ZNF335, ZNF711
Fetal (including NIPD) 125 gènes+
Conditions presenting in utero or at birth, including lethal skeletal dysplasias, fetal akinesia and arthrogryposis syndromes, hydrops fetalis, primary microcephaly, spondylocostal dysostosis, and other severe congenital conditions with prenatal onset
ADAMTSL2, ADAT3, AFG2A, AGRN, ALG2, ALX3, ANKLE2, ANTXR1, ANTXR2, ASNS, ATR, BBIP1, BIN1, BMPER, BRAT1, C1QBP, CDK10, CDK5RAP2, CDKL5, CEP135, CEP152, CEP63, CFAP418, CFL2, CHUK, CLCN4, CLCN7, COLEC10, CRLF1, CWC27, DDR2, DLL3, DNMT3B, DONSON, DPH1, DSE, EFL1, EMG1, ERBB3, FAM20A, FAT4, FTO, GLDN, GLE1, GPC6, GPX4, GZF1, HES7, HNRNPH2, HSPG2, IFT56, INPPL1, INTU, IRX5, KATNB1, LAGE3, LAMB2, LARP7, LGI4, LIFR, LNPK, LTBP4, MAP3K20, MATN3, MCPH1, MEGF10, MEOX1, MESP2, MGP, MID1, MMP13, MRPS14, MYO18B, MYO9A, MYOD1, NAA10, NANS, NHEJ1, NKX3-2, NPHS1, NUP88, PAK3, PAPSS2, PDE6D, PGAP1, PHF8, PKD1L1, PLCB4, PLG, PLPBP, POP1, PRG4, PRRX1, PSPH, RAB33B, RAD50, RAX, RBM10, ROGDI, RSPO2, SASS6, SCYL2, SLC26A3, SLC33A1, SLC35A3, SMS, SOST, SPINT2, STAG2, TBCK, TBX15, TCTN2, THOC2, TP53RK, TPRKB, TRIP11, TSEN15, TUBGCP4, TXN2, TXNL4A, UBE3B, UBR1, UNC13D, VPS51, ZNHIT3
Mitochondrial 114 gènes+
Mitochondrial respiratory chain deficiencies, mitochondrial DNA depletion and deletion syndromes, coenzyme Q10 deficiency, pyruvate metabolism disorders, mitochondrial translation defects, and other mitochondrial energy metabolism disorders
AARS2, ABAT, ACAD9, AGK, ATAD3A, ATP5F1D, ATP5MK, BCS1L, BOLA3, BTD, C2orf69, CLPB, COA6, COA8, COQ2, COQ6, COQ8B, COQ9, COX15, COX20, COX6A2, COX6B1, COX7B, COX8A, CYC1, DLD, DNA2, DNM2, EARS2, ECHS1, ELAC2, ETHE1, FARS2, FBXL4, FLAD1, GLRX5, HCCS, HIBCH, HSPA9, IBA57, ISCA1, ISCA2, KARS1, LIAS, LIPT1, LONP1, LRPPRC, LYRM7, MDH2, MFN2, MGME1, MICOS13, MICU1, MIPEP, MPV17, MRPS2, MRPS22, MTO1, MTRFR, NARS2, NDUFA1, NDUFA11, NDUFA13, NDUFA6, NDUFA9, NDUFAF2, NDUFAF3, NDUFAF5, NDUFAF6, NDUFAF8, NDUFB3, NDUFB8, NDUFS1, NDUFS2, NDUFS3, NDUFV2, NFU1, OPA1, PDHB, PDHX, PDP1, PDSS1, PDSS2, PET100, PMPCA, PMPCB, POLG2, RARS2, SARS2, SCO1, SERAC1, SFXN4, SLC19A2, SLC19A3, SLC25A19, SLC25A26, SLC25A3, SLC25A46, TARS2, TIMMDC1, TK2, TMEM126B, TPK1, TRMT5, TRMU, TRNT1, TSFM, TWNK, UQCC2, UQCRC2, UQCRFS1, VARS1, VARS2, YARS2
Musculoskeletal 113 gènes+
Osteogenesis imperfecta, skeletal dysplasias, craniosynostosis syndromes, osteopetrosis, Ehlers-Danlos syndromes, limb malformation disorders, short-rib thoracic dysplasias, and other bone and connective tissue disorders
ADAMTS2, ALPL, ALX4, AMER1, ATP6V1A, BBS10, BHLHA9, BMP1, BMP2, BMPR1B, BPNT2, C2CD3, CA2, CCN6, CCNQ, CDC45, CEP120, CFAP410, CILK1, COL11A2, COL1A2, COL27A1, COLEC11, CREB3L1, CRTAP, DLX5, DOCK6, DYNC2H1, DYNC2I1, DYNC2I2, DYNC2LI1, EFNB1, EOGT, ESCO2, EVC2, FAM20C, FGD1, FGFR3, FKBP10, FLNB, GDF5, GORAB, GPC3, HDAC8, IFT122, IFT140, IFT172, IFT80, IFT81, IHH, IL11RA, KDELR2, KIF7, LBR, LMBR1, LRP4, LRP5, LTBP3, MASP1, MEGF8, MESD, MKS1, MMP2, NBAS, NEK1, OSTM1, P3H1, PAX3, PHEX, PISD, PLOD2, PLS3, POLR1D, PPIB, PRMT7, PTH1R, RAB23, RIN2, RNU4ATAC, ROR2, RSPRY1, SCARF2, SEC23A, SEC24D, SERPINF1, SERPINH1, SHOX, SLC10A7, SLC26A2, SLC39A13, SMC1A, SNX10, SP7, SPARC, TAPT1, TBX22, TBX4, TCF12, TCIRG1, TENT5A, TGDS, TGFB1, TMCO1, TMEM38B, TNFRSF11A, TNFRSF11B, TNFSF11, TRAPPC2, TRPV6, USP9X, WDR35, WNT10B, WNT7A
Endocrinology 105 gènes+
Congenital adrenal hyperplasia, disorders of sex development, congenital hypothyroidism, hypogonadotropic hypogonadism, congenital hyperinsulinism, neonatal and monogenic diabetes, growth hormone deficiency, lipodystrophies, autoimmune polyendocrinopathy, and other endocrine disorders
ABCC8, ACAN, AGPAT2, AIRE, ALMS1, ANOS1, AQP2, AR, ARNT2, CACNA1D, CASR, CAV1, CAVIN1, CCDC8, CDT1, CEP57, CISD2, CPAP, CRIPT, CUL4B, CUL7, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP27B1, CYP2R1, DHH, DMP1, DUOX2, DUOXA2, EIF2AK3, FANCM, FEZF1, FOXE1, FOXP3, FSHB, GCK, GHR, GLIS3, GNRH1, GNRHR, HAMP, HESX1, HSD17B3, HSD3B2, IER3IP1, IGF1R, IGSF1, INS, INSR, KCNJ11, KDM6A, KISS1R, LHB, LHX3, LRBA, MAMLD1, MC2R, NEUROG3, NPR2, NR0B1, NSMCE2, OBSL1, ORC1, ORC4, ORC6, PCBD1, PCNT, PCSK1, PCYT1A, PIK3R1, PLK4, POC1A, POMC, POU1F1, PPP1R15B, PROP1, PTF1A, RBBP8, RFX6, RPL10, SAMD9, SGPL1, SLC29A3, SLC34A1, SLC34A3, SLC5A5, SOX3, SRD5A2, STAR, STAT5B, TAC3, TACR3, TBCE, TBX19, TFR2, TOP3A, TRIM37, TRMT10A, TSHB, TSHR, VDR, WFS1, ZMPSTE24
Ophthalmology 85 gènes+
Retinal dystrophies, Bardet-Biedl syndrome, Joubert syndrome with retinal involvement, congenital cataracts, microphthalmos and anophthalmos, oculocutaneous albinism, congenital glaucoma, and other hereditary eye disorders
ABCA4, ALDH1A3, ANK3, ARL13B, ARL3, ARL6, ASPH, B3GLCT, BBS1, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, BCOR, C12orf57, CC2D2A, CEP290, CEP78, CHM, CHRDL1, CLRN1, COL11A1, COL18A1, COL9A2, CRB1, CRYAA, CSPP1, FOXE3, FRAS1, FREM2, GDF6, HMX1, IGBP1, INPP5E, KIAA0586, LRP2, LTBP2, LZTFL1, MAB21L2, MFRP, MITF, MKKS, MPDZ, NDP, NHS, OFD1, PDE6G, PITX3, PLOD3, POC1B, PRDM5, PRSS56, PXDN, RAB18, RAB3GAP1, RAB3GAP2, RARB, RIC1, RIMS2, RIPK4, RPE65, RPGRIP1, RPGRIP1L, SDCCAG8, SH3PXD2B, SLC4A4, SMOC1, SRD5A3, STRA6, TBC1D20, TENM3, TMEM126A, TMEM216, TMEM231, TMEM237, TMEM67, TRAF3IP1, TTC8, TYR, TYRP1, USH1G, VPS13B, VSX2
Dermatology 67 gènes+
Epidermolysis bullosa, ichthyoses, ectodermal dysplasias, palmoplantar keratodermas, hereditary hair and nail disorders, pigmentation disorders, and other genodermatoses
ABCA12, ABHD5, ALOX12B, ALOXE3, AP1S1, CARD11, CDH3, CDSN, CERS3, COL17A1, COL7A1, CSTA, CYP4F22, DSG1, DSP, EDA, EDAR, EDARADD, EDN3, EDNRB, ENPP1, EXPH5, GJA1, GJB2, GJB3, GJB6, GRHL2, GTF2H5, HOXC13, HPGD, ITGA3, ITGA6, ITGB4, JUP, KRT10, KRT14, KRT5, LAMA3, LAMB3, LAMC2, MBTPS2, MPLKIP, MYO5A, NECTIN1, NECTIN4, NEK9, NIPAL4, PNPLA1, POMP, PORCN, RAB27A, RMRP, RSPO4, SLC27A4, SLC39A4, SMARCAL1, SNAP29, SPINK5, ST14, STS, TAT, TGM1, TSPEAR, TWIST2, USB1, UVSSA, WNT10A
Hearing 57 gènes+
Non-syndromic sensorineural hearing loss, Usher syndrome, Pendred syndrome, auditory neuropathy, and other hereditary hearing disorders
ADCY1, ADGRV1, CABP2, CDC14A, CDH23, CIB2, CLDN14, CLIC5, COCH, ELMOD3, EPS8, EPS8L2, ESPN, ESRRB, FGF3, GIPC3, GRXCR1, HGF, ILDR1, LHFPL5, LOXHD1, LRTOMT, MARVELD2, MPZL2, MSRB3, MYO15A, MYO3A, MYO6, MYO7A, OTOA, OTOF, OTOG, OTOGL, PCDH15, PDZD7, PJVK, POU3F4, PPIP5K2, PTPRQ, RDX, RIPOR2, ROR1, S1PR2, SERPINB6, SLC26A4, SLC26A5, SYNE4, TECTA, TMC1, TMIE, TMPRSS3, TPRN, TRIOBP, USH1C, USH2A, WBP2, WHRN
Immunology 57 gènes+
Severe combined immunodeficiency (SCID), agammaglobulinemias, MHC class II deficiency, chronic granulomatous disease, Mendelian susceptibility to mycobacterial disease, hemophagocytic lymphohistiocytosis, autoinflammatory syndromes, and other primary immunodeficiencies
ADA, BLNK, BTK, CARMIL2, CD19, CD247, CD27, CD3D, CD3E, CD40, CD70, CD79A, CD79B, CFP, CIITA, CORO1A, CTPS1, DOCK2, FADD, IFNGR1, IFNGR2, IKBKB, IL12RB1, IL1RN, IL21R, IL2RB, IL7R, IRAK4, IRF8, ITCH, JAK3, LAT, MALT1, MCM4, MSN, MTHFD1, MYD88, NCF4, NCKAP1L, NSMCE3, PNP, PRKCD, PRKDC, PSMB8, PTPRC, RAC2, RFX5, RFXANK, RFXAP, RIPK1, RNF168, SP110, STAT2, STX11, TAP1, TYK2, ZAP70
Cancer 55 gènes+
Fanconi anemia, xeroderma pigmentosum, dyskeratosis congenita and telomere biology disorders, hereditary breast and ovarian cancer, DNA repair deficiency syndromes, hereditary tumor predisposition syndromes, and other cancer-predisposing conditions
ACD, ATM, BLM, BRCA1, BRCA2, BUB1B, CTC1, DDB2, DIS3L2, DKC1, DOCK8, EPCAM, ERCC2, ERCC3, ERCC4, ERCC5, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FH, HAX1, ITK, LIG4, MET, MUTYH, NBN, NHP2, NOP10, PARN, POLE, PRF1, RAD51C, RECQL4, RTEL1, SDHA, SDHD, SH2D1A, SLX4, SMAD4, STN1, SUFU, TERT, TRIP13, UBE2T, WRAP53, WRN, XPA, XPC, XRCC2
Haematology 51 gènes+
Hemoglobinopathies, hereditary hemolytic anemias, congenital bleeding and coagulation disorders, bone marrow failure syndromes, congenital neutropenia, platelet disorders, thrombotic microangiopathies, and other hematological conditions
ABCB7, ADA2, ADAMTS13, AK2, AMN, AP3B1, AP3D1, ARPC1B, CDIN1, CSF3R, DIAPH1, DNAJC21, ERCC6L2, F10, F13A1, F2, F7, F8, F9, FERMT3, FGA, FGB, FGG, GATA1, GSS, HBB, HK1, JAGN1, LPIN2, MPL, MTRR, MYSM1, NT5C3A, PERCC1, PIEZO1, PKLR, PRDX1, PROC, PROS1, PUS1, RBM8A, SBDS, SLC25A38, SLC4A1, TBXAS1, TCN2, TF, TMPRSS6, TPI1, VPS45, WAS
Gastrohepatology 50 gènes+
Cystic fibrosis, progressive familial intrahepatic cholestasis, bile acid synthesis defects, congenital diarrheal disorders, very early onset inflammatory bowel disease, hereditary liver diseases, and other gastrointestinal and hepatic conditions
ABCB11, ABCB4, ADAM17, ADK, AKR1D1, ATP8B1, CD3G, CD40LG, CD55, CFTR, CLDN1, CLMP, CYBA, CYBB, DCDC2, DCLRE1C, DGAT1, FAH, FLNA, GALE, GALT, GUCY2C, HPS1, HSD3B7, ICOS, IL10RA, IL2RA, IL2RG, KRT8, MVK, MYO5B, NCF1, NCF2, NR1H4, OTULIN, RAG1, RAG2, SGO1, SKIC2, SKIC3, SLC9A3, STAT1, STXBP2, TALDO1, TJP2, TTC7A, UGT1A1, USP53, WNT2B, XIAP
Renal 46 gènes+
Hereditary nephritis, polycystic kidney disease, nephrotic syndromes, nephronophthisis, renal tubular disorders, congenital anomalies of the kidney and urinary tract, and other hereditary renal conditions
ACE, AGT, AGTR1, AHI1, ANKS6, ARHGDIA, ATP6V0A4, ATP6V1B1, BSND, CEP164, CEP83, CLCN5, CLCNKB, CLDN10, COL4A3, COL4A4, COL4A5, CYP24A1, DGKE, DSTYK, FREM1, GRIP1, HPSE2, INVS, ITGA8, KCNJ1, MAGI2, MAPKBP1, NPHP1, NPHP3, NPHP4, NPHS2, NUP107, NUP133, NUP93, OCRL, PKHD1, PLCE1, REN, SCNN1B, SCNN1G, SLC12A1, TBC1D8B, TTC21B, VIPAS39, VPS33B
Cardiology 42 gènes+
Hypertrophic and dilated cardiomyopathies, arrhythmogenic cardiomyopathy, inherited arrhythmia syndromes, hereditary aortopathies, congenital heart defects with laterality disorders, and other inherited cardiovascular conditions
ABCC6, ACTA1, ADAMTS19, AIFM1, BGN, CASQ2, CCBE1, CDH2, COL3A1, EFEMP2, EMD, FBLN5, FKBP14, GDF1, GLA, GNB5, IPO8, KCNE1, KCNQ1, LAMP2, LMNA, LZTR1, MMP21, MRPL44, MYBPC3, MYH11, MYH7, MYL3, MYPN, NAXD, NODAL, PLOD1, PPA2, PTPN14, SGCD, SGCG, SLC2A10, SPEG, TAFAZZIN, TCAP, TRDN, TSPYL1
Ciliopathies 25 gènes+
Primary ciliary dyskinesia, Meckel syndrome, short-rib polydactyly syndromes, cranioectodermal dysplasia, mucociliary clearance disorders, and other ciliopathy-related conditions
B9D2, CCNO, CEP104, DNAH5, DRC1, DRC2, DRC4, EVC, IFT27, IFT43, IFT52, IFT74, IQCB1, KIAA0753, NEK8, PIBF1, PIK3CD, RSPH1, RSPH3, SCNN1A, TMEM107, TMEM138, TXNDC15, WDPCP, WDR19
Respiratory 21 gènes+
Primary ciliary dyskinesia with respiratory manifestations, surfactant metabolism dysfunction, hereditary pulmonary alveolar proteinosis, heritable pulmonary arterial hypertension, and other respiratory conditions
ABCA3, CCDC39, CCDC40, CFAP298, CFAP300, CSF2RB, DNAAF11, DNAAF19, DNAAF3, DNAAF4, DNAAF5, DNAAF6, DNAH11, DNAH9, EIF2AK4, LRRC56, MARS1, ODAD1, ODAD2, SFTPB, SPAG1
Other 39 gènes+
Miscellaneous rare monogenic disorders not fitting a single organ-system category, including isolated congenital conditions, ultra-rare syndromes, and other genetic conditions
AIPL1, AP1B1, ATOH7, ATP6V1E1, BANF1, CANT1, CCDC47, ELMO2, ELP2, FOXN1, FRMPD4, FTSJ1, HERC1, IGFBP7, IQSEC1, KLHL15, KRT18, LINS1, MED23, METTL23, MLPH, NMNAT1, NTNG2, NUP62, NYX, PIGY, PREPL, PRSS12, SELENOI, SYP, TAF13, TNFRSF13B, TSPAN7, TTI2, UPF3B, UQCRQ, WNT3, WNT4, ZIC3
Questions fréquentes
Qu’est-ce que le dépistage de porteur (carrier screening) ?+
Le dépistage de porteur est un test génétique qui permet de déterminer si vous portez un variant génétique associé à certaines maladies héréditaires. Les porteurs sont généralement en bonne santé et ne présentent aucun symptôme. Dans la plupart des cas, il est impossible de savoir sans test de quelles maladies on est porteur.
Lorsque deux partenaires sont tous deux porteurs de la même maladie, il existe généralement, à chaque grossesse, un risque de 25 % d’avoir un enfant atteint de cette maladie. Le dépistage de porteur donne aux couples cette information avant ou pendant la grossesse afin qu’ils puissent prendre des décisions éclairées et en toute autonomie.
Combien cela coûte-t-il ?+
Le test génétique lui-même coûte 1 495 CHF et est payé à titre privé. Les séances de conseil génétique coûtent entre 200 et 300 CHF chacune et sont couvertes par l’assurance maladie de base.
Que puis-je faire avec mes résultats ?+
Si les deux partenaires sont identifiés comme porteurs de la même maladie, votre conseiller·ère en génétique vous expliquera l’ensemble des options qui s’offrent à vous. Celles-ci peuvent inclure :
- Diagnostic génétique préimplantatoire (PGT) — analyse génétique d’embryons conçus par FIV avant l’établissement d’une grossesse
- Diagnostic prénatal pendant la grossesse — comme la biopsie des villosités choriales (CVS) ou l’amniocentèse pour analyser directement la grossesse
- Poursuivre avec cette information — certains couples choisissent de continuer une grossesse en connaissant pleinement les issues possibles
- Conception avec donneur ou adoption — des options que certains couples porteurs envisagent
Il n’existe pas de réponse unique « juste », et notre équipe est là pour vous accompagner dans la voie qui vous convient. Prenez rendez-vous pour discuter de votre situation particulière.
Le contenu médical de deena.health est révisé par Dr Isis Atallah, spécialiste FMH en Génétique médicale. Dernière révision le .