
Gentest für die
Familienplanung
Trägerabklärung auf 2'000+ Erbkrankheiten
Für CHF 1'495
Bequem von zu Hause aus.
Warum ein Gentest sinvoll ist
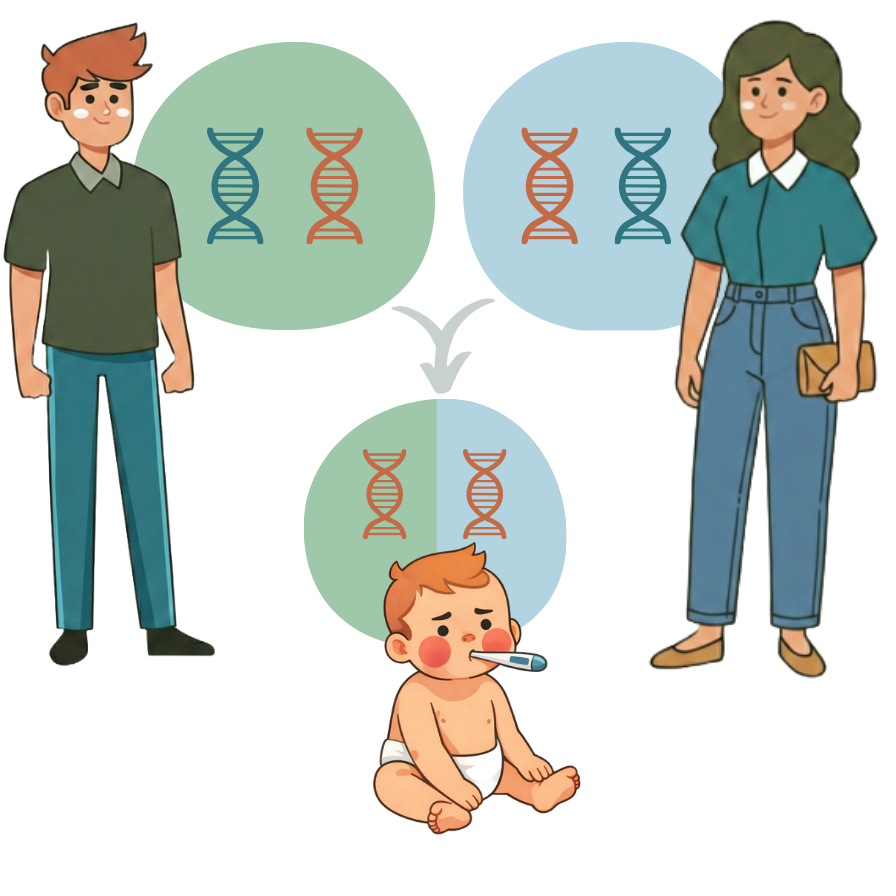
Für viele Eltern überraschend haben die meisten Babys, die mit bestimmten genetischen Erkrankungen geboren werden, gesunde Eltern, die selbst nicht davon betroffen sind. Der Grund dafür ist, dass viele Menschen Träger genetischer Mutationen sind, ohne es zu wissen. Diese Mutationen können dazu führen, dass ein Gen defekt ist. Da wir aber von den meisten Genen zwei Kopien haben (eine von jedem Elternteil), kompensiert eine gesunde Kopie die defekte. Wenn jedoch beide Eltern zufällig Träger derselben rezessiven Erkrankung sind, besteht ein Risiko von 25 %, dass ihr Kind beide defekten Kopien erbt und betroffen sein wird.
Grosse Studien haben gezeigt, wie verbreitet diese erblichen genetischen Erkrankungen in der Allgemeinbevölkerung sind. In einer US-Studie mit Hunderttausenden von Personen lag der Anteil betroffener Schwangerschaften bei etwa 1 von 175 [1]. Ebenso wurden in einem landesweiten australischen Screening-Programm mit über zehntausend Paaren etwa 1 von 50 Paaren als Risikopaar identifiziert [2].
- Westemeyer M, Saucier J, Wallace J, et al. Clinical experience with carrier screening in a general population: support for a comprehensive pan-ethnic approach. Genetics in Medicine. 2020;22:1320–1328. doi:10.1038/s41436-020-0807-4
- Mackenzie's Mission Investigators. Nationwide implementation of reproductive genetic carrier screening in Australia. New England Journal of Medicine. 2024. doi:10.1056/NEJMoa2314768
Der Test
Der Gentest zur Abklärung der Trägerschaft ist ein DNA-basierter Test, der das genetische Material beider Partner analysiert, um festzustellen, ob sie gemeinsam Träger bestimmter Erbkrankheiten sind. Mithilfe der Next-Generation-Sequenzierung können dabei gleichzeitig Hunderte bis Tausende von Genen untersucht werden. Unser Test analysiert gezielt 1’944 Gene, die mit schweren Erkrankungen assoziiert sind, die Kinder typischerweise früh im Leben betreffen. Der Ablauf ist einfach und nicht-invasiv und basiert auf einer Speichelprobe, aus der DNA gewonnen und sequenziert wird. Der Test konzentriert sich auf rezessive und X-chromosomale Erkrankungen — genetische Störungen, die bei Eltern in der Regel keine Symptome verursachen, aber bei ihren Kindern zu schweren Erkrankungen führen können, wenn beide Partner dieselbe Erkrankung tragen oder bestimmte Vererbungsmuster vorliegen.
Das Labor
Wir arbeiten mit der CeGaT GmbH als Partnerlabor zusammen. CeGaT führt die genetische Analyse mit einem eigens entwickelten Assay durch, der sowohl Einzelnukleotidvarianten als auch Kopienzahlvarianten detektiert und gezielte Tests für Erkrankungen wie das Fragile-X-Syndrom und die spinale Muskelatrophie umfasst. Das Labor verfügt über wichtige internationale Akkreditierungen für klinische Gentests, darunter CAP, CLIA und ISO 15189.
Genetische Beratung
Ein wesentlicher Bestandteil unseres Angebots ist die genetische Beratung vor und nach dem Test durch eine Fachperson. Diese Sitzungen bieten die Möglichkeit, Fragen zu stellen, den Umfang und die Aussagekraft des Tests zu verstehen und die Ergebnisse im Detail zu besprechen. Durch die Überprüfung der persönlichen und familiären Vorgeschichte hilft die Beratung, Befunde in den Kontext einzuordnen und fundierte Entscheidungen zu unterstützen.
So funktioniert es
Den eigenen Trägerstatus bequem zu Hause testen.
Genetische Beratung
In einem Online-Gespräch mit einer Fachperson für Genetik erfahren, was der Gentest für die Familienplanung bedeutet.
Speichelprobe entnehmen
Eine einfache Speichelprobe zu Hause mit dem zugesendeten Kit entnehmen. Kein Arztbesuch nötig — einfach versiegeln und mit dem beiliegenden Rücksendeetikett zurückschicken.
Ergebnisse besprechen
Die Ergebnisse mit einer Fachärztin oder einem Facharzt besprechen und verstehen, was die Befunde für Ihre Familienplanung bedeuten.
Genliste
Neurology 423 Gene+
Muscular dystrophies, congenital myopathies, congenital myasthenic syndromes, hereditary motor and sensory neuropathies, epileptic encephalopathies, leukodystrophies, neuronal ceroid lipofuscinoses, hereditary ataxias, hereditary spastic paraplegias, brain malformations, peroxisomal disorders, neurodegeneration with brain iron accumulation, and other neuromuscular and neurodegenerative conditions
AAAS, AARS1, ABCD1, ABHD12, ACOX1, ACP5, ADAR, ADCY5, ADCY6, ADGRG1, ADGRG6, ADPRS, AFG3L2, AGTPBP1, AIMP1, ALDH3A2, ALDH5A1, ALG13, ALS2, AMPD2, ANO5, AP1S2, AP3B2, AP4M1, AP4S1, APC2, APTX, ARFGEF2, ARL6IP1, ARMC9, ARSA, ARV1, ARX, ASCC1, ASPA, ASPM, ATCAY, ATP13A2, ATP1A2, ATP2B3, ATP7B, AVIL, B3GALNT2, B4GALNT1, B4GAT1, BCAP31, BLTP1, BRF1, BSCL2, C19orf12, CA8, CAPN3, CASK, CCT5, CENPF, CEP41, CERS1, CHAT, CHMP1A, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, CLCN1, CLCN2, CLP1, CNPY3, CNTNAP1, COASY, COL13A1, COL6A1, COL6A2, COL6A3, COLQ, COQ4, COQ8A, COX10, CPLANE1, CRB2, CRPPA, CRYAB, CSF1R, CSTB, CTDP1, CTNNA2, CWF19L1, CYP27A1, CYP2U1, CYP7B1, D2HGDH, DAG1, DARS1, DARS2, DCAF17, DCX, DDC, DDHD1, DDHD2, DDX59, DENND5A, DGUOK, DHDDS, DLAT, DMD, DMXL2, DNAJC19, DNAJC3, DNAJC6, DOK7, DPYD, DST, DYSF, ECEL1, EGR2, EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S3, ELOVL4, ELP1, EML1, ENTPD1, EPM2A, ERCC6, ERCC8, ERLIN1, ERLIN2, ETFDH, EXOC3L2, EXOSC3, EXOSC8, EXOSC9, FA2H, FAM149B1, FBXO7, FGD4, FHL1, FIG4, FKRP, FKTN, FLVCR1, FLVCR2, FOLR1, FRRS1L, FTL, FUCA1, FXN, GALC, GBA2, GBE1, GCH1, GJC2, GMPPB, GOLGA2, GOSR2, GOT2, GPAA1, GPSM2, GPT2, GRID2, GRIN1, GRM1, GRM7, GUCY1A1, GUF1, HACD1, HACE1, HADHA, HADHB, HARS1, HEPACAM, HEXA, HEXB, HIKESHI, HINT1, HPDL, HSD17B10, HSPD1, HYCC1, HYLS1, IFIH1, IGHMBP2, INPP5K, ITGA7, ITPR1, JAM2, JAM3, KCNJ10, KCNMA1, KDM5C, KIDINS220, KIF1A, KIF1C, KIFBP, KLHL40, KLHL41, KY, L2HGDH, LAMA2, LAMB1, LAMC3, LARGE1, LIMS2, LMOD3, LPIN1, LYST, MAG, MBOAT7, MECP2, MECR, MLC1, MMACHC, MPZ, MRE11, MSTO1, MTFMT, MTM1, MTMR2, MTTP, MUSK, MYBPC1, MYH3, MYMK, NARS1, NAXE, NCAPD3, NDE1, NDRG1, NDUFA12, NDUFA2, NDUFS4, NDUFS7, NDUFS8, NDUFV1, NEB, NFASC, NGF, NHLRC1, NKX6-2, NRROS, NRXN1, NT5C2, NTRK1, NUP188, OCLN, OPA3, OPHN1, ORAI1, OSGEP, PANK2, PCCA, PCCB, PCDH12, PCYT2, PDE10A, PDHA1, PEX1, PEX10, PEX12, PEX13, PEX16, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7, PI4KA, PIEZO2, PIP5K1C, PLA2G6, PLCB1, PLEC, PLEKHG5, PLP1, PMP22, PNKP, PNPLA6, PNPT1, POLG, POLR3A, POLR3B, POMGNT1, POMGNT2, POMK, POMT1, POMT2, PRDM12, PRICKLE1, PRKRA, PRPS1, PRX, PTPN23, PTRH2, PTS, PYCR2, PYROXD1, QDPR, RAB39B, RAD21, RAPSN, RARS1, RELN, RETREG1, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2, ROBO3, RRM2B, RTTN, RXYLT1, RYR1, SACS, SAMHD1, SBF1, SBF2, SCARB2, SCN1B, SCN4A, SCN9A, SCO2, SELENON, SEPSECS, SETX, SGCA, SGCB, SH3TC2, SIL1, SLC12A6, SLC16A2, SLC18A3, SLC1A4, SLC25A12, SLC25A4, SLC2A1, SLC30A10, SLC39A14, SLC52A2, SLC52A3, SLC5A7, SLC6A3, SLC6A5, SLC6A8, SLC9A1, SLC9A6, SMPD4, SNORD118, SNX14, SOD1, SPART, SPG11, SPR, SPTBN2, SPTBN4, SQSTM1, STAC3, STAMBP, STIM1, STRADA, STUB1, SUCLA2, SUOX, SURF1, SVBP, SYN1, SYNE1, SYNJ1, SZT2, TANGO2, TBC1D24, TDP2, TECPR2, TH, TIMM8A, TMX2, TNNT1, TOE1, TPM3, TRAK1, TRAPPC11, TRAPPC12, TREX1, TRIM2, TRIM32, TRIP4, TSEN2, TSEN54, TTC19, TTN, TTPA, TYMP, UBA1, UBA5, UCHL1, UFC1, UGP2, UNC80, USP18, VAC14, VAMP1, VLDLR, VMA21, VPS11, VPS13D, VPS37A, VPS41, VPS53, VRK1, WARS2, WASHC5, WDR45, WDR45B, WDR62, WDR73, WDR81, WNK1, WWOX, ZC4H2, ZFYVE26
Metabolic 286 Gene+
Inborn errors of metabolism including amino acid disorders, organic acidemias, fatty acid oxidation disorders, urea cycle disorders, glycogen storage diseases, lysosomal storage disorders, peroxisomal disorders, congenital disorders of glycosylation, cholesterol biosynthesis defects, mineral and metal metabolism disorders, and other metabolic conditions
ABCD4, ACADM, ACADS, ACADSB, ACADVL, ACAT1, ACOX2, ACY1, ADSL, AGA, AGL, AGPS, AGXT, ALAD, ALDH18A1, ALDH4A1, ALDH6A1, ALDH7A1, ALDOA, ALDOB, ALG1, ALG11, ALG12, ALG14, ALG3, ALG6, ALG8, AMACR, AMPD1, AMT, ANO10, ARG1, ARSB, ARSL, ASAH1, ASL, ASS1, ATIC, ATP6AP1, ATP6V0A2, ATP7A, ATPAF2, AUH, B3GALT6, B3GAT3, B4GALT1, B4GALT7, BCKDHA, BCKDHB, BCKDK, CA5A, CARS2, CBS, CHST14, CHST3, CHSY1, CLDN16, CLDN19, CLN3, CLN5, CLN6, CLN8, CLPP, CNNM2, COG1, COG2, COG4, COG5, COG6, COG7, COQ7, COX14, CPS1, CPT1A, CPT2, CTNS, CTSA, CTSD, CTSK, DHCR24, DHCR7, DHODH, DHTKD1, DNAJC12, DNM1L, DOLK, DPAGT1, DPM1, DPM2, DYM, EBP, EPG5, ERAL1, ETFA, ETFB, FAR1, FASTKD2, FBP1, FCSK, FITM2, FTCD, FUT8, G6PC1, G6PC3, GAA, GALK1, GALNS, GAMT, GBA1, GCDH, GDAP1, GFER, GFM1, GFM2, GFPT1, GK, GLB1, GLDC, GLS, GLYCTK, GM2A, GMPPA, GNPAT, GNPTAB, GNPTG, GNS, GPHN, GRHPR, GTPBP3, GUSB, GYS1, GYS2, HADH, HARS2, HCFC1, HGSNAT, HJV, HLCS, HMGCL, HMGCS2, HOGA1, HPRT1, HSD11B2, HSD17B4, HTRA2, HYAL1, IARS2, IDS, IDUA, ITPA, IVD, KCTD7, LARS2, LDHA, LDLR, LFNG, LIPA, LMBRD1, LPL, LYRM4, MAGT1, MAN1B1, MAN2B1, MANBA, MAOA, MAT1A, MCCC1, MCCC2, MCEE, MCOLN1, MFSD8, MGAT2, MLYCD, MMAA, MMAB, MMADHC, MMUT, MOCS1, MOCS2, MOGS, MPDU1, MPI, MRPL3, MRPS16, MSMO1, MTHFR, NAGA, NAGLU, NAGS, NDUFA10, NDUFAF1, NDUFAF4, NDUFS6, NEU1, NGLY1, NNT, NPC1, NPC2, NSDHL, NUBPL, OGDH, OTC, OXCT1, PAH, PARS2, PC, PCK1, PEPD, PEX11B, PEX14, PEX19, PFKM, PGAP2, PGAP3, PGK1, PGM1, PGM3, PHGDH, PHKG2, PHYH, PIGA, PIGL, PIGN, PIGO, PIGS, PIGT, PIGV, PMM2, PNPO, POR, PPT1, PRODH, PSAP, PSAT1, PYCR1, PYGL, PYGM, RBCK1, RFT1, RMND1, SAR1B, SC5D, SEC23B, SGSH, SLC12A3, SLC16A1, SLC17A5, SLC22A5, SLC25A13, SLC25A15, SLC25A20, SLC25A22, SLC25A42, SLC2A2, SLC35A1, SLC35A2, SLC35C1, SLC35D1, SLC37A4, SLC39A8, SLC3A1, SLC46A1, SLC5A1, SLC7A7, SMPD1, SSR4, ST3GAL3, STT3A, SUCLG1, SUMF1, TACO1, TMEM165, TMEM70, TPP1, TRMT10C, TRPM6, TUFM, TUSC3, UMPS, UQCRB, UROS, VMA12, VMA22, VPS33A, XYLT1, XYLT2
Developmental 179 Gene+
Syndromic and non-syndromic intellectual disability, global developmental delay, autism spectrum-associated syndromes, brain malformations, GPI anchor deficiency syndromes, DNA repair and chromosome maintenance disorders, and other neurodevelopmental conditions
ABCC9, ACACA, ACO2, ACSL4, ACTL6B, ADAM22, ADARB1, AFF2, AHCY, AIMP2, ALG9, AP4B1, AP4E1, ARHGEF9, ATAD1, ATG7, ATP6AP2, ATP8A2, ATRX, B9D1, BRWD3, CAD, CAMK2A, CC2D1A, CCDC22, CCDC88A, CCDC88C, CDH11, CEP55, CHKB, CIT, CKAP2L, CNKSR2, CNTNAP2, CPLX1, CRADD, CRBN, CTU2, CYB5R3, DBT, DCHS1, DDX11, DDX3X, DEAF1, DEGS1, DHX37, DLG3, DOCK7, EIF4A3, EMC1, EMC10, EPRS1, ERCC1, EXT2, EXTL3, FOXL2, FOXRED1, GAD1, GAN, GATM, GCSH, GDI1, GEMIN4, GLUL, GRIA3, GRIK2, HERC2, HOXA1, HPD, HUWE1, IARS1, IGF1, IL1RAPL1, INTS1, IQSEC2, KATNIP, KDM5B, KIF14, KLHL7, KNL1, KPTN, L1CAM, LAMA1, LAS1L, MED12, MED17, MED25, METTL5, MFSD2A, MRPS34, MSL3, MTR, NADSYN1, NALCN, NDST1, NECAP1, NEMF, NEXMIF, NKAP, NONO, NSUN2, NUDT2, OTUD5, OTUD6B, OXR1, PAM16, PCDH19, PHF6, PIGB, PIGG, PIGK, PIGP, PIGQ, PLAA, PLEKHG2, PLVAP, PNPLA8, POLA1, POLR1C, PPP1R21, PQBP1, PRUNE1, PTCHD1, PUS7, QARS1, RALGAPA1, RDH11, RINT1, RLIM, RNF113A, RNF13, RPIA, RPS6KA3, RTN4IP1, RUSC2, SCAPER, SCYL1, SDHAF1, SLC12A5, SLC13A5, SLC25A1, SLC5A6, SLC6A9, ST3GAL5, STIL, TAF1, TAF2, TAF6, TASP1, TBC1D23, TBCD, TCTN3, TELO2, THOC6, TIMM50, TMEM132E, TMEM260, TMEM94, TMTC3, TRAIP, TRAPPC4, TRAPPC9, TRIT1, TRMT1, TUBGCP2, TUBGCP6, UBE2A, UFM1, UGDH, UROC1, WDR4, WNT1, XRCC4, YIF1B, ZBTB24, ZC3H14, ZDHHC9, ZNF335, ZNF711
Fetal (including NIPD) 125 Gene+
Conditions presenting in utero or at birth, including lethal skeletal dysplasias, fetal akinesia and arthrogryposis syndromes, hydrops fetalis, primary microcephaly, spondylocostal dysostosis, and other severe congenital conditions with prenatal onset
ADAMTSL2, ADAT3, AFG2A, AGRN, ALG2, ALX3, ANKLE2, ANTXR1, ANTXR2, ASNS, ATR, BBIP1, BIN1, BMPER, BRAT1, C1QBP, CDK10, CDK5RAP2, CDKL5, CEP135, CEP152, CEP63, CFAP418, CFL2, CHUK, CLCN4, CLCN7, COLEC10, CRLF1, CWC27, DDR2, DLL3, DNMT3B, DONSON, DPH1, DSE, EFL1, EMG1, ERBB3, FAM20A, FAT4, FTO, GLDN, GLE1, GPC6, GPX4, GZF1, HES7, HNRNPH2, HSPG2, IFT56, INPPL1, INTU, IRX5, KATNB1, LAGE3, LAMB2, LARP7, LGI4, LIFR, LNPK, LTBP4, MAP3K20, MATN3, MCPH1, MEGF10, MEOX1, MESP2, MGP, MID1, MMP13, MRPS14, MYO18B, MYO9A, MYOD1, NAA10, NANS, NHEJ1, NKX3-2, NPHS1, NUP88, PAK3, PAPSS2, PDE6D, PGAP1, PHF8, PKD1L1, PLCB4, PLG, PLPBP, POP1, PRG4, PRRX1, PSPH, RAB33B, RAD50, RAX, RBM10, ROGDI, RSPO2, SASS6, SCYL2, SLC26A3, SLC33A1, SLC35A3, SMS, SOST, SPINT2, STAG2, TBCK, TBX15, TCTN2, THOC2, TP53RK, TPRKB, TRIP11, TSEN15, TUBGCP4, TXN2, TXNL4A, UBE3B, UBR1, UNC13D, VPS51, ZNHIT3
Mitochondrial 114 Gene+
Mitochondrial respiratory chain deficiencies, mitochondrial DNA depletion and deletion syndromes, coenzyme Q10 deficiency, pyruvate metabolism disorders, mitochondrial translation defects, and other mitochondrial energy metabolism disorders
AARS2, ABAT, ACAD9, AGK, ATAD3A, ATP5F1D, ATP5MK, BCS1L, BOLA3, BTD, C2orf69, CLPB, COA6, COA8, COQ2, COQ6, COQ8B, COQ9, COX15, COX20, COX6A2, COX6B1, COX7B, COX8A, CYC1, DLD, DNA2, DNM2, EARS2, ECHS1, ELAC2, ETHE1, FARS2, FBXL4, FLAD1, GLRX5, HCCS, HIBCH, HSPA9, IBA57, ISCA1, ISCA2, KARS1, LIAS, LIPT1, LONP1, LRPPRC, LYRM7, MDH2, MFN2, MGME1, MICOS13, MICU1, MIPEP, MPV17, MRPS2, MRPS22, MTO1, MTRFR, NARS2, NDUFA1, NDUFA11, NDUFA13, NDUFA6, NDUFA9, NDUFAF2, NDUFAF3, NDUFAF5, NDUFAF6, NDUFAF8, NDUFB3, NDUFB8, NDUFS1, NDUFS2, NDUFS3, NDUFV2, NFU1, OPA1, PDHB, PDHX, PDP1, PDSS1, PDSS2, PET100, PMPCA, PMPCB, POLG2, RARS2, SARS2, SCO1, SERAC1, SFXN4, SLC19A2, SLC19A3, SLC25A19, SLC25A26, SLC25A3, SLC25A46, TARS2, TIMMDC1, TK2, TMEM126B, TPK1, TRMT5, TRMU, TRNT1, TSFM, TWNK, UQCC2, UQCRC2, UQCRFS1, VARS1, VARS2, YARS2
Musculoskeletal 113 Gene+
Osteogenesis imperfecta, skeletal dysplasias, craniosynostosis syndromes, osteopetrosis, Ehlers-Danlos syndromes, limb malformation disorders, short-rib thoracic dysplasias, and other bone and connective tissue disorders
ADAMTS2, ALPL, ALX4, AMER1, ATP6V1A, BBS10, BHLHA9, BMP1, BMP2, BMPR1B, BPNT2, C2CD3, CA2, CCN6, CCNQ, CDC45, CEP120, CFAP410, CILK1, COL11A2, COL1A2, COL27A1, COLEC11, CREB3L1, CRTAP, DLX5, DOCK6, DYNC2H1, DYNC2I1, DYNC2I2, DYNC2LI1, EFNB1, EOGT, ESCO2, EVC2, FAM20C, FGD1, FGFR3, FKBP10, FLNB, GDF5, GORAB, GPC3, HDAC8, IFT122, IFT140, IFT172, IFT80, IFT81, IHH, IL11RA, KDELR2, KIF7, LBR, LMBR1, LRP4, LRP5, LTBP3, MASP1, MEGF8, MESD, MKS1, MMP2, NBAS, NEK1, OSTM1, P3H1, PAX3, PHEX, PISD, PLOD2, PLS3, POLR1D, PPIB, PRMT7, PTH1R, RAB23, RIN2, RNU4ATAC, ROR2, RSPRY1, SCARF2, SEC23A, SEC24D, SERPINF1, SERPINH1, SHOX, SLC10A7, SLC26A2, SLC39A13, SMC1A, SNX10, SP7, SPARC, TAPT1, TBX22, TBX4, TCF12, TCIRG1, TENT5A, TGDS, TGFB1, TMCO1, TMEM38B, TNFRSF11A, TNFRSF11B, TNFSF11, TRAPPC2, TRPV6, USP9X, WDR35, WNT10B, WNT7A
Endocrinology 105 Gene+
Congenital adrenal hyperplasia, disorders of sex development, congenital hypothyroidism, hypogonadotropic hypogonadism, congenital hyperinsulinism, neonatal and monogenic diabetes, growth hormone deficiency, lipodystrophies, autoimmune polyendocrinopathy, and other endocrine disorders
ABCC8, ACAN, AGPAT2, AIRE, ALMS1, ANOS1, AQP2, AR, ARNT2, CACNA1D, CASR, CAV1, CAVIN1, CCDC8, CDT1, CEP57, CISD2, CPAP, CRIPT, CUL4B, CUL7, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP27B1, CYP2R1, DHH, DMP1, DUOX2, DUOXA2, EIF2AK3, FANCM, FEZF1, FOXE1, FOXP3, FSHB, GCK, GHR, GLIS3, GNRH1, GNRHR, HAMP, HESX1, HSD17B3, HSD3B2, IER3IP1, IGF1R, IGSF1, INS, INSR, KCNJ11, KDM6A, KISS1R, LHB, LHX3, LRBA, MAMLD1, MC2R, NEUROG3, NPR2, NR0B1, NSMCE2, OBSL1, ORC1, ORC4, ORC6, PCBD1, PCNT, PCSK1, PCYT1A, PIK3R1, PLK4, POC1A, POMC, POU1F1, PPP1R15B, PROP1, PTF1A, RBBP8, RFX6, RPL10, SAMD9, SGPL1, SLC29A3, SLC34A1, SLC34A3, SLC5A5, SOX3, SRD5A2, STAR, STAT5B, TAC3, TACR3, TBCE, TBX19, TFR2, TOP3A, TRIM37, TRMT10A, TSHB, TSHR, VDR, WFS1, ZMPSTE24
Ophthalmology 85 Gene+
Retinal dystrophies, Bardet-Biedl syndrome, Joubert syndrome with retinal involvement, congenital cataracts, microphthalmos and anophthalmos, oculocutaneous albinism, congenital glaucoma, and other hereditary eye disorders
ABCA4, ALDH1A3, ANK3, ARL13B, ARL3, ARL6, ASPH, B3GLCT, BBS1, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, BCOR, C12orf57, CC2D2A, CEP290, CEP78, CHM, CHRDL1, CLRN1, COL11A1, COL18A1, COL9A2, CRB1, CRYAA, CSPP1, FOXE3, FRAS1, FREM2, GDF6, HMX1, IGBP1, INPP5E, KIAA0586, LRP2, LTBP2, LZTFL1, MAB21L2, MFRP, MITF, MKKS, MPDZ, NDP, NHS, OFD1, PDE6G, PITX3, PLOD3, POC1B, PRDM5, PRSS56, PXDN, RAB18, RAB3GAP1, RAB3GAP2, RARB, RIC1, RIMS2, RIPK4, RPE65, RPGRIP1, RPGRIP1L, SDCCAG8, SH3PXD2B, SLC4A4, SMOC1, SRD5A3, STRA6, TBC1D20, TENM3, TMEM126A, TMEM216, TMEM231, TMEM237, TMEM67, TRAF3IP1, TTC8, TYR, TYRP1, USH1G, VPS13B, VSX2
Dermatology 67 Gene+
Epidermolysis bullosa, ichthyoses, ectodermal dysplasias, palmoplantar keratodermas, hereditary hair and nail disorders, pigmentation disorders, and other genodermatoses
ABCA12, ABHD5, ALOX12B, ALOXE3, AP1S1, CARD11, CDH3, CDSN, CERS3, COL17A1, COL7A1, CSTA, CYP4F22, DSG1, DSP, EDA, EDAR, EDARADD, EDN3, EDNRB, ENPP1, EXPH5, GJA1, GJB2, GJB3, GJB6, GRHL2, GTF2H5, HOXC13, HPGD, ITGA3, ITGA6, ITGB4, JUP, KRT10, KRT14, KRT5, LAMA3, LAMB3, LAMC2, MBTPS2, MPLKIP, MYO5A, NECTIN1, NECTIN4, NEK9, NIPAL4, PNPLA1, POMP, PORCN, RAB27A, RMRP, RSPO4, SLC27A4, SLC39A4, SMARCAL1, SNAP29, SPINK5, ST14, STS, TAT, TGM1, TSPEAR, TWIST2, USB1, UVSSA, WNT10A
Hearing 57 Gene+
Non-syndromic sensorineural hearing loss, Usher syndrome, Pendred syndrome, auditory neuropathy, and other hereditary hearing disorders
ADCY1, ADGRV1, CABP2, CDC14A, CDH23, CIB2, CLDN14, CLIC5, COCH, ELMOD3, EPS8, EPS8L2, ESPN, ESRRB, FGF3, GIPC3, GRXCR1, HGF, ILDR1, LHFPL5, LOXHD1, LRTOMT, MARVELD2, MPZL2, MSRB3, MYO15A, MYO3A, MYO6, MYO7A, OTOA, OTOF, OTOG, OTOGL, PCDH15, PDZD7, PJVK, POU3F4, PPIP5K2, PTPRQ, RDX, RIPOR2, ROR1, S1PR2, SERPINB6, SLC26A4, SLC26A5, SYNE4, TECTA, TMC1, TMIE, TMPRSS3, TPRN, TRIOBP, USH1C, USH2A, WBP2, WHRN
Immunology 57 Gene+
Severe combined immunodeficiency (SCID), agammaglobulinemias, MHC class II deficiency, chronic granulomatous disease, Mendelian susceptibility to mycobacterial disease, hemophagocytic lymphohistiocytosis, autoinflammatory syndromes, and other primary immunodeficiencies
ADA, BLNK, BTK, CARMIL2, CD19, CD247, CD27, CD3D, CD3E, CD40, CD70, CD79A, CD79B, CFP, CIITA, CORO1A, CTPS1, DOCK2, FADD, IFNGR1, IFNGR2, IKBKB, IL12RB1, IL1RN, IL21R, IL2RB, IL7R, IRAK4, IRF8, ITCH, JAK3, LAT, MALT1, MCM4, MSN, MTHFD1, MYD88, NCF4, NCKAP1L, NSMCE3, PNP, PRKCD, PRKDC, PSMB8, PTPRC, RAC2, RFX5, RFXANK, RFXAP, RIPK1, RNF168, SP110, STAT2, STX11, TAP1, TYK2, ZAP70
Cancer 55 Gene+
Fanconi anemia, xeroderma pigmentosum, dyskeratosis congenita and telomere biology disorders, hereditary breast and ovarian cancer, DNA repair deficiency syndromes, hereditary tumor predisposition syndromes, and other cancer-predisposing conditions
ACD, ATM, BLM, BRCA1, BRCA2, BUB1B, CTC1, DDB2, DIS3L2, DKC1, DOCK8, EPCAM, ERCC2, ERCC3, ERCC4, ERCC5, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FH, HAX1, ITK, LIG4, MET, MUTYH, NBN, NHP2, NOP10, PARN, POLE, PRF1, RAD51C, RECQL4, RTEL1, SDHA, SDHD, SH2D1A, SLX4, SMAD4, STN1, SUFU, TERT, TRIP13, UBE2T, WRAP53, WRN, XPA, XPC, XRCC2
Haematology 51 Gene+
Hemoglobinopathies, hereditary hemolytic anemias, congenital bleeding and coagulation disorders, bone marrow failure syndromes, congenital neutropenia, platelet disorders, thrombotic microangiopathies, and other hematological conditions
ABCB7, ADA2, ADAMTS13, AK2, AMN, AP3B1, AP3D1, ARPC1B, CDIN1, CSF3R, DIAPH1, DNAJC21, ERCC6L2, F10, F13A1, F2, F7, F8, F9, FERMT3, FGA, FGB, FGG, GATA1, GSS, HBB, HK1, JAGN1, LPIN2, MPL, MTRR, MYSM1, NT5C3A, PERCC1, PIEZO1, PKLR, PRDX1, PROC, PROS1, PUS1, RBM8A, SBDS, SLC25A38, SLC4A1, TBXAS1, TCN2, TF, TMPRSS6, TPI1, VPS45, WAS
Gastrohepatology 50 Gene+
Cystic fibrosis, progressive familial intrahepatic cholestasis, bile acid synthesis defects, congenital diarrheal disorders, very early onset inflammatory bowel disease, hereditary liver diseases, and other gastrointestinal and hepatic conditions
ABCB11, ABCB4, ADAM17, ADK, AKR1D1, ATP8B1, CD3G, CD40LG, CD55, CFTR, CLDN1, CLMP, CYBA, CYBB, DCDC2, DCLRE1C, DGAT1, FAH, FLNA, GALE, GALT, GUCY2C, HPS1, HSD3B7, ICOS, IL10RA, IL2RA, IL2RG, KRT8, MVK, MYO5B, NCF1, NCF2, NR1H4, OTULIN, RAG1, RAG2, SGO1, SKIC2, SKIC3, SLC9A3, STAT1, STXBP2, TALDO1, TJP2, TTC7A, UGT1A1, USP53, WNT2B, XIAP
Renal 46 Gene+
Hereditary nephritis, polycystic kidney disease, nephrotic syndromes, nephronophthisis, renal tubular disorders, congenital anomalies of the kidney and urinary tract, and other hereditary renal conditions
ACE, AGT, AGTR1, AHI1, ANKS6, ARHGDIA, ATP6V0A4, ATP6V1B1, BSND, CEP164, CEP83, CLCN5, CLCNKB, CLDN10, COL4A3, COL4A4, COL4A5, CYP24A1, DGKE, DSTYK, FREM1, GRIP1, HPSE2, INVS, ITGA8, KCNJ1, MAGI2, MAPKBP1, NPHP1, NPHP3, NPHP4, NPHS2, NUP107, NUP133, NUP93, OCRL, PKHD1, PLCE1, REN, SCNN1B, SCNN1G, SLC12A1, TBC1D8B, TTC21B, VIPAS39, VPS33B
Cardiology 42 Gene+
Hypertrophic and dilated cardiomyopathies, arrhythmogenic cardiomyopathy, inherited arrhythmia syndromes, hereditary aortopathies, congenital heart defects with laterality disorders, and other inherited cardiovascular conditions
ABCC6, ACTA1, ADAMTS19, AIFM1, BGN, CASQ2, CCBE1, CDH2, COL3A1, EFEMP2, EMD, FBLN5, FKBP14, GDF1, GLA, GNB5, IPO8, KCNE1, KCNQ1, LAMP2, LMNA, LZTR1, MMP21, MRPL44, MYBPC3, MYH11, MYH7, MYL3, MYPN, NAXD, NODAL, PLOD1, PPA2, PTPN14, SGCD, SGCG, SLC2A10, SPEG, TAFAZZIN, TCAP, TRDN, TSPYL1
Ciliopathies 25 Gene+
Primary ciliary dyskinesia, Meckel syndrome, short-rib polydactyly syndromes, cranioectodermal dysplasia, mucociliary clearance disorders, and other ciliopathy-related conditions
B9D2, CCNO, CEP104, DNAH5, DRC1, DRC2, DRC4, EVC, IFT27, IFT43, IFT52, IFT74, IQCB1, KIAA0753, NEK8, PIBF1, PIK3CD, RSPH1, RSPH3, SCNN1A, TMEM107, TMEM138, TXNDC15, WDPCP, WDR19
Respiratory 21 Gene+
Primary ciliary dyskinesia with respiratory manifestations, surfactant metabolism dysfunction, hereditary pulmonary alveolar proteinosis, heritable pulmonary arterial hypertension, and other respiratory conditions
ABCA3, CCDC39, CCDC40, CFAP298, CFAP300, CSF2RB, DNAAF11, DNAAF19, DNAAF3, DNAAF4, DNAAF5, DNAAF6, DNAH11, DNAH9, EIF2AK4, LRRC56, MARS1, ODAD1, ODAD2, SFTPB, SPAG1
Other 39 Gene+
Miscellaneous rare monogenic disorders not fitting a single organ-system category, including isolated congenital conditions, ultra-rare syndromes, and other genetic conditions
AIPL1, AP1B1, ATOH7, ATP6V1E1, BANF1, CANT1, CCDC47, ELMO2, ELP2, FOXN1, FRMPD4, FTSJ1, HERC1, IGFBP7, IQSEC1, KLHL15, KRT18, LINS1, MED23, METTL23, MLPH, NMNAT1, NTNG2, NUP62, NYX, PIGY, PREPL, PRSS12, SELENOI, SYP, TAF13, TNFRSF13B, TSPAN7, TTI2, UPF3B, UQCRQ, WNT3, WNT4, ZIC3
Häufig gestellte Fragen
Was ist die Abklärung einer Anlageträgerschaft (Carrier-Screening)?+
Die Abklärung einer Anlageträgerschaft (Carrier-Screening) ist ein Gentest, der feststellt, ob man eine Genvariante trägt, die mit bestimmten Erbkrankheiten verbunden ist. Träger sind in der Regel gesund und haben keine Symptome. In den meisten Fällen ist es ohne Test unmöglich zu wissen, für welche Erkrankungen man ein Träger ist.
Wenn beide Partner Träger derselben Erkrankung sind, besteht bei jeder Schwangerschaft in der Regel eine Wahrscheinlichkeit von 25 %, dass das Kind von dieser Erkrankung betroffen ist. Der Gentest gibt Paaren diese Information vor oder während der Schwangerschaft, damit sie informierte Entscheidungen treffen können.
Wie viel kostet der Test?+
Der Gentest selbst kostet CHF 1'495.- und wird privat bezahlt. Genetische Beratungssitzungen kosten je CHF 200–300.- und werden von der regulären Krankenversicherung übernommen.
Was macht man mit den Testergebnissen?+
Wenn beide Partner als Träger derselben Erkrankung identifiziert werden, erläutert die genetische Beraterin oder der Berater die gesamte Bandbreite der verfügbaren Optionen. Dazu gehören unter anderem:
- Präimplantationsdiagnostik (PGT) — genetische Untersuchung von Embryonen, die durch IVF erzeugt wurden, bevor eine Schwangerschaft eintritt
- Pränataldiagnostik während der Schwangerschaft — wie Chorionzottenbiopsie (CVS) oder Amniozentese zur direkten Untersuchung des ungeborenen Kindes
- Fortfahren mit diesen Informationen — manche Paare entscheiden sich, eine Schwangerschaft mit vollem Wissen über die möglichen Ergebnisse fortzusetzen
- Samenspende oder Adoption — Optionen, die manche Träger-Paare in Betracht ziehen
Es gibt keine einzig «richtige» Antwort, und unser Team unterstützt Sie auf dem Weg, der sich für Sie richtig anfühlt. Vereinbaren Sie eine Beratung, um Ihre spezifische Situation zu besprechen.
Medizinische Inhalte auf deena.health werden von Dr. Isis Atallah, FMH Medizinische Genetik, geprüft. Zuletzt überprüft am .